Structural and dynamical properties of organic materials are investigated by first-principles molecular dynamics (FPMD). In this approach, we resort to the electronic structure calculated with the density-functional theory (DFT) to obtain the interatomic forces and achieve a predictive modeling at finite temperature, of the thermal properties for example. We use the code cpmd.
Exciton diffusion
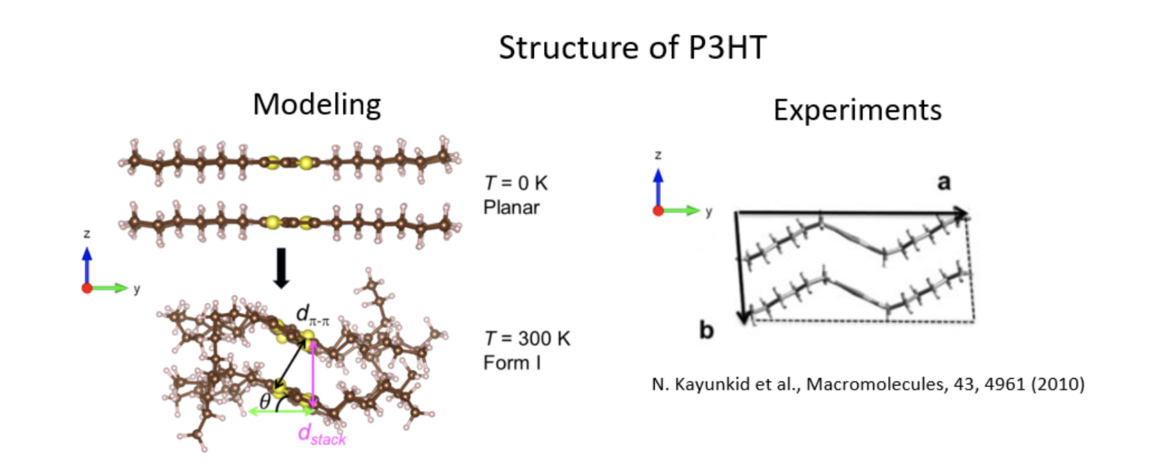
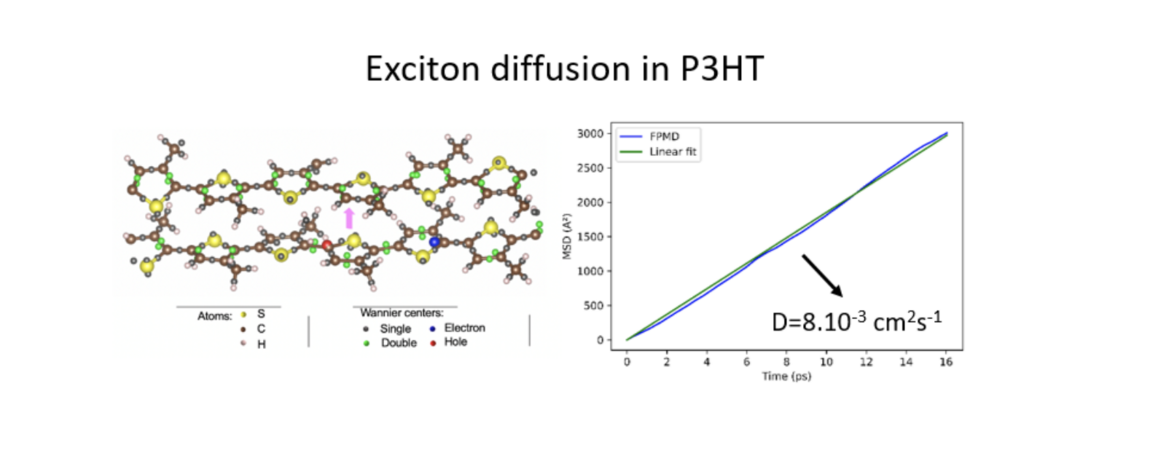
A key parameter in organic solar cells is the exciton diffusion length, that drives the efficiency of the device since a larger length enables the photo-generated electron-hole pair to reach the donor-acceptor interface where they are dissociated to be collected. As a first step, we target the exciton diffusion coefficient. The S1 excited state is modeled within the ROKS (Restricted-Open shell Kohn-Sham) formalism. We resort to the Wannier functions centers (WFCs) to localize the excitonon the fly from the combined motion of ions and orbitals. In the case of the P3HT polymer, we have recently shown that our approach enables to describe the so-called Form I of the P3HT crystals.
Moreover, by calculating the mean square displacement of the exciton WFCs, we were able to calculate the diffusion coefficient along the p-p stacking direction, and obtained an excellent agreement with experimental measurements on regio-regular P3HT. The result paves the way to an atomic-level optimization of this key parameter on the computer. See our recent publication in Phys. Chem. Chem. Phys.!
Thermal properties
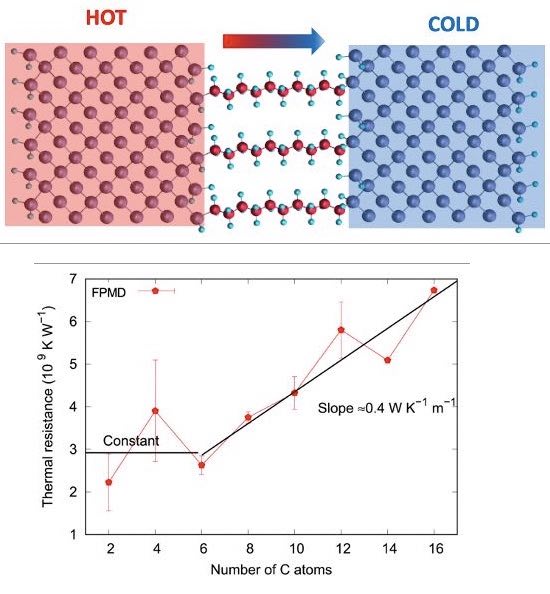
One of our achievements was to study the thermal transport through a layer of molecules :
The transient time that characterizes the approach-to-equilibrium (uniform temperature in the system) is used to obtain the thermal interface resistance of the molecular layer. When the calculation was repeated with different lengths of the molecules, we were able to distinguish a constant contribution coming from the bond between the reservoirs and the molecules, and a diffusive contribution with a thermal conductivity of 0.4 W K-1m-1.